Your free guide to current MDR Classification Rules
GuideThe classification of medical devices has changed since the implementation of the EU MDR in May 2021. To ensure you’re meeting the latest classification rules, we’ve put together a handy infographic and complete guide covering the 22 MDR rules across standard medical devices and in-vitro diagnostic devices.
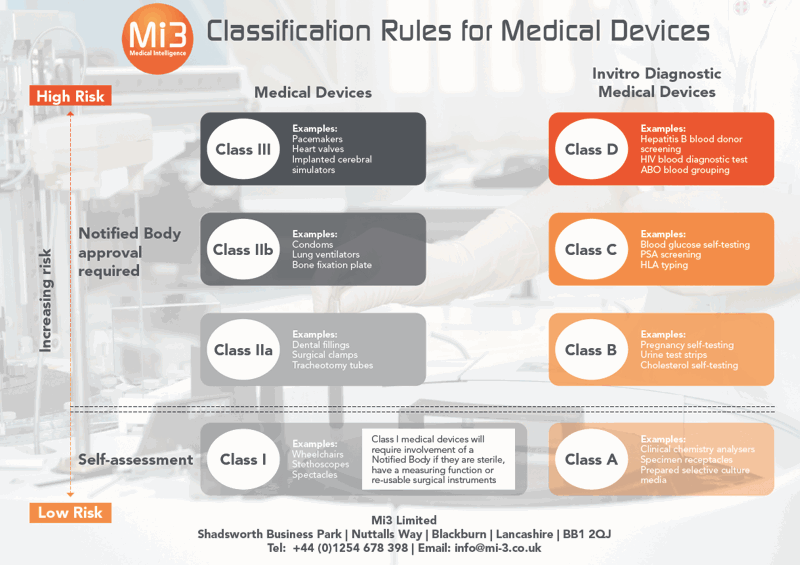
This infographic offers a snapshot of the key changes, but to get the full picture of the classification updates – and to see if your existing products require re-classifying – download our comprehensive 8-page copy of the essential MDR Classification Rules Guide…
Our guide will provide clarification on whether your existing products require re-classifying, and ensure you know exactly what any new devices should be classified as, too. The MDR Classification Rules Guide covers:
- The classifications for non-invasive devices
- The classifications for invasive devices
- The classifications for active devices
- Special rules for devices
- The background on understanding the changes
- Who the classification rules apply to
Previous Post Next Post